Höchstleistungsrechenzentrum Stuttgart
„Mit der Verbesserung der rechnergestützten Methoden in den letzten 40 Jahren sind Berechnungen zu einem wichtigen Hilfsmittel für die Biophysik und die pharmazeutische Forschung geworden“, sagt Dr. Giovanni Settanni, der an der Ruhr-Universität Bochum und der Johannes Gutenberg-Universität Mainz (JGU) in computergestützter Biophysik forscht. „Simulationen können als eine Art In-silico-Mikroskop mit einer Auflösung auf molekularer Ebene angesehen werden. Sie helfen bei der Interpretation experimenteller Daten, die kaum die gleiche Auflösung erreichen können.“
Settanni ist Experte für Molekulardynamiksimulation (MD). Dieser Forschungsbereich entwickelt anhand von Prinzipien aus der Biophysik Modelle von Molekülen auf atomarer Ebene. MD-Methoden werden in der biomedizinischen Forschung u.a. zur Vorhersage von Veränderungen in der Proteinstruktur eingesetzt. Solche Veränderungen wirken sich auf die Fähigkeit eines Proteins aus, mit anderen Molekülen zu interagieren und sich an sie zu binden. Wegen ihrer extrem hohen Auflösung und weil sie zeigen, wie sich die Form von Proteinen im Laufe der Zeit verändern kann, können MD-Simulationen nur mit Supercomputern durchgeführt werden. Kürzlich zeigte Settani, wie sich anhand von Simulationen auf dem Supercomputer Hawk am Höchstleistungsrechenzentrum Stuttgart (HLRS) neue Behandlungsmöglichkeiten gegen Krebs entdecken lassen.
Dr. Settanni hat mit der Gruppe des Strukturbiologen Dr. Andreas Joerger von der Goethe-Universität Frankfurt zusammengearbeitet, um ein wichtiges Protein namens p53 zu untersuchen. In einem gesunden menschlichen Körper dient p53 als Bollwerk gegen Krebs, indem es reguliert, wie sich Zellen teilen und wie sie DNA-Schäden reparieren. Wenn jedoch eine Mutation in p53 entsteht, kann sich wegen einer Veränderung des genetischen Codes die Aminosäuresequenz ändern, die auch die 3D-Struktur des Proteins beeinflusst. Dies kann dazu führen, dass das Protein seine normalen regulatorischen Fähigkeiten verliert. Beim Ausfall dieser Bremse können sich Krebszellen unkontrolliert teilen und ausbreiten. Etwa die Hälfte aller Krebsfälle geht auf Mutationen zurück, die das p53-Protein deaktivieren.
Bei einem Ansatz in der Krebsforschung suchen Wissenschaftler:innen nach Medikamenten, die schädliche Mutationen neutralisieren können, indem sie sich mit Proteinen verbinden. Im Fall von p53 suchen sie nach kleinen Molekülen, die die Mutationen stabilisieren und die ursprüngliche Form des Proteins wiederherstellen können. Wenn ein geeignetes Molekül gefunden würde, könnte p53 wieder normal funktionieren. Bis vor Kurzem hielten Krebsforschende p53 jedoch für unangreifbar, da seine Oberfläche für die Bindung mit kleinen Molekülen praktisch überall ungeeignet zu sein schien.
In früheren Forschungsarbeiten zeigte Joerger, dass es möglich sei, eine bestimmte Mutation namens Y220C zu behandeln. Die Y220C-Mutation verursacht eine spezifische physikalische Veränderung, die einen destabilisierenden Spalt in der Oberfläche von p53 erzeugt. Die Veränderung führt dazu, dass das Protein bei Körpertemperatur seine normale Form verliert. Sein Labor identifizierte ein kleines Molekül, das sich an die Oberfläche des Y220C-Hohlraums bindet und so p53 wieder in seine richtige Form bringt. Im Labor zeigten die Forschenden auch, dass der Ansatz die Anti-Krebs-Funktion des Proteins wiederherstellt. Dieses Ergebnis führte zu einem klinischen Versuch, um die Sicherheit und Wirksamkeit dieser Strategie am Menschen zu testen.
Angesichts dieses Erfolgs sucht Joerger nun nach anderen potenziell behandelbaren Mutationen in p53. Er möchte auch im Allgemeinen verstehen, ob mutationsspezifische, kleine Moleküle einen wirksamen Weg zur Wiederherstellung der Funktion des kritischen Proteins bieten. Hier leisten Simulationen nun einen wichtigen Beitrag.
Früher arbeitete Joerger hauptsächlich mit Röntgenkristallografie im Labor, um die Proteinstruktur zu verstehen. Diese Technologie funktioniert wie ein hochauflösendes Mikroskop, bei dem ein kristallisiertes Protein bestrahlt wird, um eine Momentaufnahme auf atomarer Ebene zu erstellen. Kürzlich hat er jedoch mit Settanni zusammengearbeitet, um Molekulardynamiksimulationen in seine Forschung einzubeziehen. MD-Simulationen ergänzen die Röntgenkristallographie, indem sie ein umfassenderes und detaillierteres Verständnis dafür liefern, wie sich die Form von Proteinen im Laufe der Zeit verändern kann. Der Ansatz kann neue Einblicke in das Verhalten eines Proteins und in seine Anfälligkeit für Medikamente unter verschiedenen Bedingungen liefern.
Wie in einem Artikel in der Fachzeitschrift Cell Death and Disease berichtet, untersuchte das Team eine bestimmte Klasse von strukturell instabilen, krebsverursachenden Mutationen in p53. In diesem Zusammenhang nutzte Settanni den Supercomputer Hawk des HLRS, um mehrere Simulationen verschiedener Mutanten von p53 durchzuführen. Die Simulationen, die auch das umgebende Lösungsmittel einschließen, enthalten zwischen 36.000 und 39.000 Atome und ermöglichen es, die zeitliche Entwicklung jeder Mutante kumulativ für 0,8 Mikrosekunden zu verfolgen.
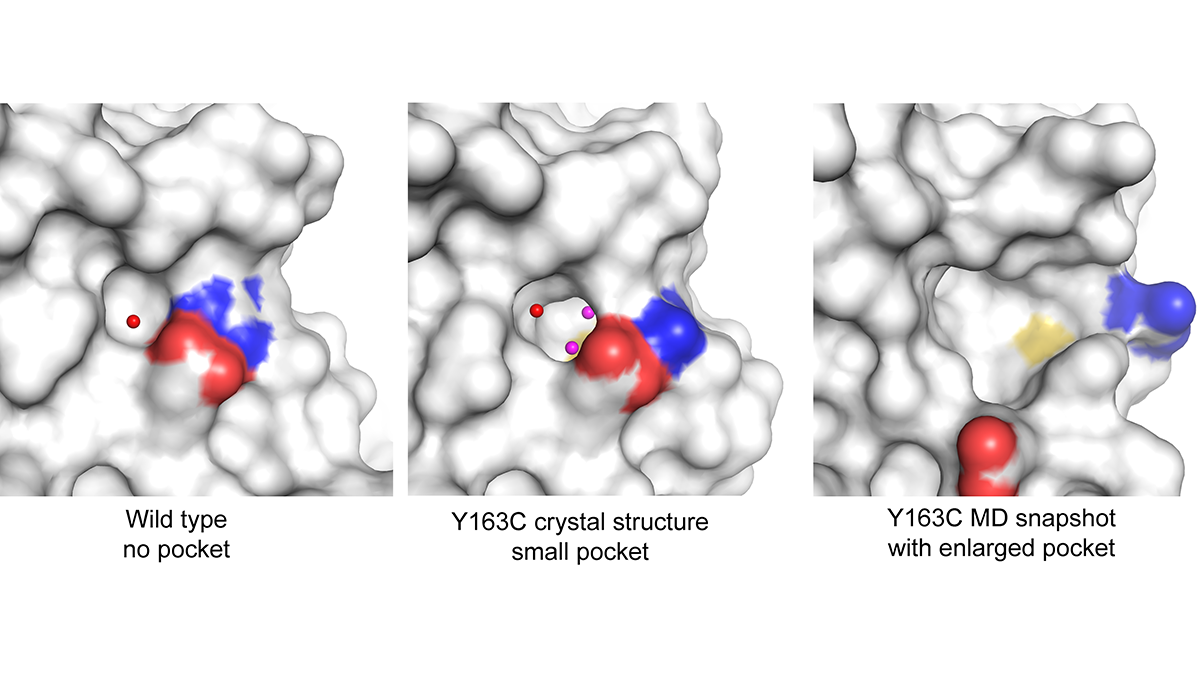
„Simulationen halfen, das Verhalten der weniger stabilen Mutanten von p53 zu charakterisieren, die experimentell schwer zu handhaben sind“, so Settanni. „Simulationen können das Vorhandensein von Hohlräumen identifizieren, die sich auf der Oberfläche des mutierten Proteins bilden können. Diese Hohlräume können mit Medikamenten angegriffen werden, um das Protein zu stabilisieren.“
Laut der Studie erwies sich eine Mutation namens Y163C als besonders interessant. Am Anfang deutete ein Röntgenkristallographie-Bild darauf hin, dass Y163C kein gutes Ziel sein würde, weil der durch die Mutation entstandene Hohlraum sehr klein ist. MD-Simulationen zeigten jedoch, dass sich die Form dieses Hohlraums drastisch vergrößern lässt, was reichlich Platz für ein kleines Molekül zur Bindung an das Protein bietet. Nach dieser Erkenntnis konzentrierten sich die Forschenden auf Y163C statt auf andere Mutationen, die laut MD und anderen Ansätzen resistenter gegen die Bindung sind.
Die MD-Simulationen lieferten auch strukturelle Details des Y163C-Hohlraums, die bei der Suche nach bindefähigen kleinen Molekülen für p53 helfen könnten. Diese Informationen können das virtuelle Wirkstoffscreening, einen weiteren Ansatz in der computergestützten Biologie, unterstützen. Hier werden Datenkataloge, die viele tausend verschiedene Arten von kleinen Molekülen beschreiben, mit den molekularen und strukturellen Merkmalen einer potenziellen Bindungsstelle verglichen. Anhand eines virtuellen Screenings kann schnell festgestellt werden, welche kleinen Moleküle am ehesten kompatibel sind. Dieser Ansatz spart viel Zeit und Arbeit, da nun nicht mehr jede potenzielle Interaktion im Labor zu testen ist. Für die relativ geringe Anzahl an kleinen Molekülen, die mit hoher Wahrscheinlichkeit mit p53 interagieren, können anschließend in Laborexperimenten schnell die rechnerischen Vorhersagen untersucht werden. Wenn diese Experimente letztendlich erfolgreich sind, ließen sich klinische Versuche mit den Ergebnissen rechtfertigen. Forschende könnten dann untersuchen, ob die Verwendung eines kleinen Moleküls zur Bekämpfung von Y163C in Menschen sicher und wirksam ist.
Es gibt keine Garantie dafür, dass sich Y163C als wirksames Zielmolekül zur Bekämpfung von Krebs erweist. Die Zusammenarbeit zwischen Joerger und Settanni zeigt jedoch, wie Molekulardynamiksimulationen mithilfe von Höchstleistungsrechnern einen wichtigen Beitrag zur Krebsforschung leisten und den Fortschritt in der personalisierten Krebsmedizin beschleunigen können.
— Christopher Williams
Balourdas DI, Markl AM, Krämer A, et al. 2024. Structural basis of p53 inactivation by cavity-creating cancer mutations and its implications for the development of mutant p53 reactivators. Cell Death Dis. 15: 408.
Hawk wurde vom Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg und vom Bundesministerium für Bildung und Forschung über das Gauss Centre for Supercomputing (GCS) finanziert.
Als Mitglied des Gauss Centre for Supercomputing ist das HLRS eines der drei Bundeshöchstleistungsrechenzentren.