Höchstleistungsrechenzentrum Stuttgart

Über Ammoniak ist vieles bekannt, doch das Interesse an seiner Verwendung als Brennstoff hat die Suche nach neuen Ammoniaktechnologien ausgelöst. Dies hat wiederum zu einem erhöhten Bedarf an genauen Daten zur Beschreibung der grundlegenden thermodynamischen Eigenschaften von Ammoniak geführt. Zu diesen Eigenschaften gehört eine Vielzahl von messbaren Merkmalen wie Phasengleichgewichte, Dichte oder Wärmekapazität, die physikalische Systeme charakterisieren und die Funktionsweise chemischer Prozesse bestimmen. Ingenieur:innen möchten auch besser verstehen, wie sich diese Eigenschaften verändern, wenn Ammoniak mit anderen Molekülen gemischt wird. Dieses Wissen könnte ihnen dabei helfen, Prozesse und Betriebsbedingungen zu optimieren.
Dr. Jadran Vrabec, derzeit Direktor des Instituts für Prozesswissenschaften an der Technischen Universität Berlin, hat einen Großteil seiner beruflichen Laufbahn damit verbracht, thermodynamische Eigenschaften auf molekularer Ebene mithilfe von Höchstleistungsrechnern (HPC) zu untersuchen. „Thermodynamische Eigenschaften werden zu 100 Prozent von molekularen Wechselwirkungen bestimmt“, erklärt er. „Weil diese Wechselwirkungen so schnell und auf einer so kleinen Skala ablaufen, lassen sie sich nur mithilfe von großen Simulationen mit Supercomputern untersuchen.“
In einer kürzlich im Journal of Chemical & Engineering Data veröffentlichten Arbeit berichten er und sein Mitautor, Erich Mace von der TU Berlin, über die Ergebnisse von Simulationen, die sich auf die thermodynamischen Eigenschaften von ammoniakhaltigen Gemischen konzentrieren. Die mit dem Supercomputer Hawk am Höchstleistungsrechenzentrum Stuttgart (HLRS) erstellten Ergebnisse liefern wertvolle Daten, die die Entwicklung neuer Anwendungen von Ammoniak unterstützen könnten. Die Resultate tragen auch zur Bewertung der Genauigkeit anderer Daten bei, um sicherzustellen, dass Ingenieur:innen über die passenden Informationen für die Arbeit mit dieser Substanz verfügen.
Vrabec ist langjähriger Nutzer der Supercomputerressourcen des HLRS für Molekulardynamik- und Monte-Carlo-Simulationen. Sein Ansatz beruht auf Konzepten der Thermodynamik, die erstmals von Ludwig Boltzmann im 19. Jahrhundert formuliert wurden, aber erst in den 1950er Jahren mit dem Aufkommen der ersten Computer praktisch anwendbar wurden. Seitdem hat sich das Gebiet parallel zur Entwicklung größerer und schnellerer Supercomputer weiterentwickelt, sodass Vrabecs Simulationen heute die individuellen Bewegungen und Wechselwirkungen von Milliarden oder sogar Billionen von Molekülen gleichzeitig verfolgen. Mithilfe der von seinem Labor entwickelten Software zur selektiven Erfassung von relevanten Daten kann er die thermodynamischen Eigenschaften der Moleküle untersuchen.
Vrabec verwendet zwei Simulationscodes namens ms2 und ls1, die er im Laufe einer langen und fruchtbaren Zusammenarbeit mit den HLRS-Mitarbeitern Martin Bernreuther und Christoph Niethammer entwickelt und optimiert hat. Im Jahr 2019 stellte das Team sogar einen Weltrekord für das größte jemals mit Molekulardynamikmethoden simulierte System auf. Mit ls1 skalierten sie ihren Code effizient auf ein System mit 21 Billionen Atomen, in dem jedes einzelne Molekül und seine Wechselwirkungen mit anderen Molekülen verfolgt werden konnte.
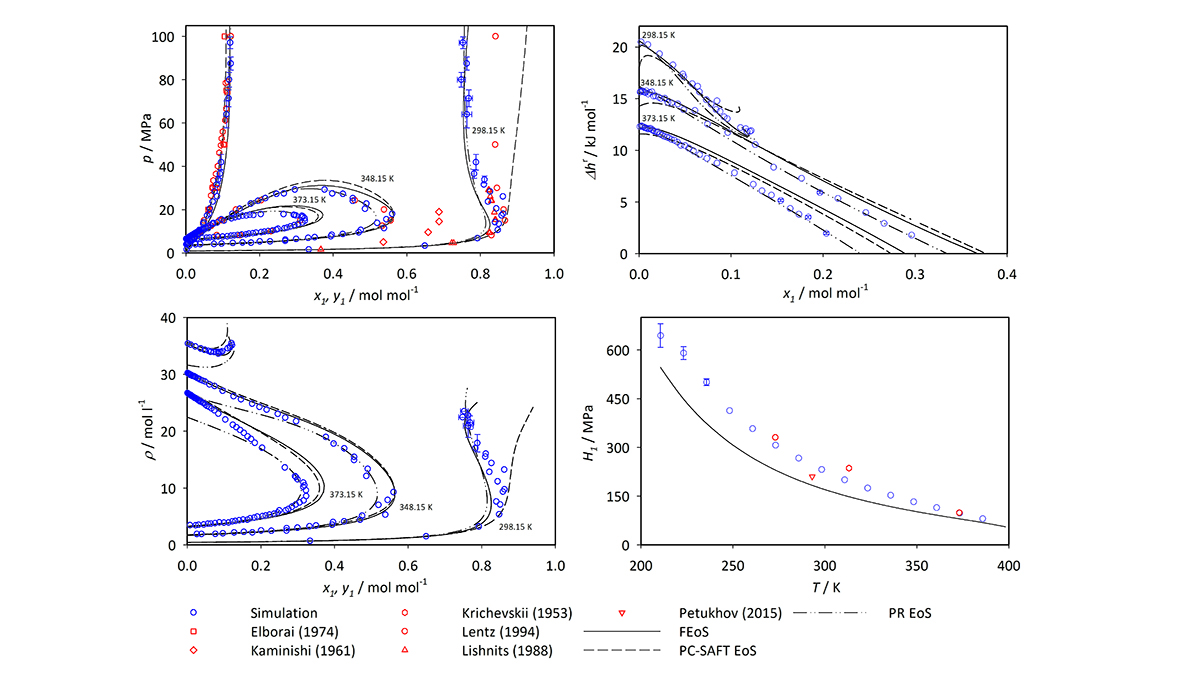
In ihrer aktuellen Arbeit über Ammoniak führten Mace und Vrabec Molekulardynamik- und Monte-Carlo-Simulationen mit ms2 durch, um fünf häufig verwendete Mischungen zu untersuchen, die Ammoniak in chemisch-technischen Prozessen enthalten: Argon-Ammoniak, Methan-Ammoniak, Wasserstoff-Ammoniak, Stickstoff-Ammoniak und Sauerstoff-Ammoniak. Für jedes Gemisch wurden bei den Simulationen Daten erzeugt, die das Vapor-Liquid-Equilibrium (VLE) – eine Messung der Verteilung der Moleküle in einem System über die Dampf- oder Flüssigphase – für einen breiten Temperatur- und Druckbereich beschreiben. In ihrem Beitrag weisen Mace und Vrabec darauf hin, dass VLE-Daten häufig bei der Entwicklung von Zustandsgleichungen für industrielle Flüssigkeiten verwendet werden, d.h. die Daten lassen sich zur Vorhersage des Zustands der Materie unter verschiedenen physikalischen Bedingungen aufgrund von Änderungen der Temperatur, des Drucks, des Volumens oder der Zusammensetzung nutzen. Solche Informationen sind für die Bestimmung optimaler Mischungen und Arbeitsbedingungen in industriellen Anwendungen unerlässlich.
Vrabecs Molekülsimulationen sind besonders wertvoll, weil mit ihnen ein viel größerer Bereich von Skalen untersucht werden kann als mit experimentellen Ansätzen. „In unseren Simulationen konnten wir die thermodynamischen Eigenschaften sogar bis zu einem Druck von 50 Megapascal messen. Das ist das 500-fache unseres Luftdrucks“, so Vrabec. „Obwohl seit mehr als einem Jahrhundert Daten für Ammoniakgemische gesammelt werden, ist die Datenabdeckung erstaunlich gering. Der Aufwand für eine experimentelle Messung ist schlichtweg unerschwinglich. Dazu wären teure und gefährliche Spezialgeräte erforderlich. Mit Computersimulationen können wir sicher und relativ kostengünstig Ergebnisse erzielen.“ Außerdem bieten seine Methoden in Bereichen, in denen experimentelle Daten verfügbar sind, ein vergleichbares Maß an Genauigkeit wie experimentelle Ansätze.
Bei der Analyse ihrer Simulationsdaten stellten Mace und Vrabec fest, dass Ammoniak zwar in allen fünf untersuchten Systemen eine Komponente ist; die Ergebnisgraphen der VLE-Werte sehen jedoch für verschiedene Molekülmischungen sehr unterschiedlich aus. Dazu Vrabec: „Das Phasenverhalten verschiedener Gemische wird stark von den Wechselwirkungen zwischen den Molekülen im System bestimmt. Man muss diese Eigenschaften verstehen, wenn man sich für die Arbeit mit Ammoniakmischungen interessiert.“
Die Forschungsarbeit und ergänzende Daten bieten mehr als 400 neue Datenpunkte für jedes untersuchte Gemisch. Mit Hawk erhielten sie die Ergebnisse für alle Gemische in jeweils nur wenigen Tagen Rechenzeit. Die Resultate sind besonders wertvoll für extreme Bedingungen, für die nur wenige Daten zur Verfügung stehen. Sie könnten Ingenieur:innen helfen, die optimalen Bedingungen für eine effiziente Ammoniakverarbeitung zu ermitteln.
Die Studie umfasste sowohl neue Simulationsdaten als auch bereits veröffentlichte Daten, sodass Mace und Vrabec ihre Ergebnisse mit anderen vorhandenen Datensätzen von VLE-Werten vergleichen konnten. In den meisten Fällen stimmten ihre Ergebnisse eng mit denen früherer Studien überein. In einigen Fällen stellten sie jedoch erhebliche Abweichungen zwischen ihren Ergebnissen und den experimentellen Messungen sowie Vorhersagen anderer Forschergruppen fest. Die Autoren führen diese Diskrepanzen auf Einschränkungen oder Ungenauigkeiten bei den entsprechenden Experimenten zurück. Sie empfehlen außerdem, dass bestimmte experimentelle Datenquellen in der künftigen Forschung oder bei verfahrenstechnischen Anwendungen mit Vorsicht verwendet werden sollten.
Vrabec hat sich kürzlich vor allem mit der Simulation thermodynamischer Eigenschaften molekularer Systeme im Submikrometerbereich beschäftigt. Auch wenn zwischen dieser Skala und beobachtbaren Prozessen einige Größenordnungen liegen, lassen sich diese Erkenntnisse mit genauen Methoden auf molekularer Ebene Vorhersagen für die Realität treffen. Vrabec geht jedoch davon aus, dass mit zunehmend größeren Supercomputern sowohl Eigenschaften als auch thermodynamische Prozesse unter realitätsnahen Randbedingungen simulierbar werden. Eine gesteigerte HPC-Leistung könnte genauere Ergebnisse über dynamische Phänomene mit einem besseren Signal-Rausch-Verhältnis liefern.
In der Zwischenzeit unterstreichen die Ergebnisse seines Teams jedoch den Wert der Molekulardynamik und der Monte-Carlo-Simulation mithilfe von Höchstleistungsrechnern. Sie werden Erkenntnisse über das Phasenverhalten liefern, die Ingenieur:innen zur Entwicklung von Technologien auf Ammoniakbasis nutzen können.
— Christopher Williams
Mace EJ, Vrabec J. 2024. High-pressure fluid-phase equilibria and Henry’s constants of supercritical gases in ammonia. J Chem Eng Data. 69(2): 573-589. (ePub Oct 2, 2023)
Thipev N, Seckler S, Heinen M, et al. 2019. TweTriS: twenty trillion-atom simulation. Int J High Perform C. 33(5): 838-854.
Hawk wurde vom Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg und vom Bundesministerium für Bildung und Forschung über das Gauss Centre for Supercomputing (GCS) gefördert.
Als Mitglied des Gauss Centre for Supercomputing ist das HLRS eines der drei Bundeshöchstleistungsrechenzentren.